
【入門】ISO13485とは?ISO9001・QMS省令との違いや要求事項を解説

FAQISO13485に関するよくある質問
ISO13485って何の規格ですか?
ISO13485は、医療機器に関する品質マネジメントシステム規格です。


ISO13485はどうして作られたんですか?
世界の医療機器法令規制の整合性を促進することを目的に制定されました。
ISO9001と同じなの?
ISO9001の2015年版とは要求事項の形式が異なっています。
「取引先からISO13485の取得を求められた」「医療機器市場への参入を見据え、ISO認証を検討している」そのような背景から、情報収集を始めている企業ご担当者様も多いのではないでしょうか。
医療機器メーカーや医療機器製造販売業者にとって、品質体制の整備は競争力と信頼性を左右する重要なテーマです。その中核となるのが、医療機器分野に特化した品質マネジメントシステム規格であるISO13485です。
そこで、この記事ではISO13485の基本的な概要から対象となる企業・製品、要求事項、取得メリットまで、わかりやすく解説します。
目次
ISO13485とは?

ISO13485とは、「医療機器に関する品質マネジメントシステムの規格」です。
医療機器メーカーや医療機器製造販売業者が、安全で安定した製品を継続的に提供するための体制づくりを定めています。
品質マネジメント規格として広く知られるISO9001が「組織の業務効率化や品質向上を重視している」のに対し、ISO13485は「世界の医療機器法規制との整合性を確保すること」を目的として制定されました。
医療機器分野では、製品品質はもちろんのこと、患者の安全確保が最優先事項です。そのため、組織に適したマネジメントシステムを整えるだけでなく、「法令遵守を確実に実現する」ことが求められています。
ISO13485の取得企業が増加している背景
近年、ISO13485の認証を取得する企業が増加しています。
その大きな理由に「医療機器のグローバル化」があり、輸出先での規制適合や取引先からISO13485の取得を求められるようになってきたことが挙げられます。
各国にはそれぞれ独自の医療機器規制があり、求められる規制要求事項も異なります。その中でISO13485は、各国の制度と整合しやすい品質マネジメントシステムとして国際的に認知されています。つまり、ISO13485取得により、「自社製品・サービスの安全性」を日本だけでなく海外の取引先に示すことができるのです。
そのため、取引先からの要請や海外展開を見据えて、取得を検討する企業が増えているのです。

ISO13485の対象となる製品・企業

ISO13485の対象となる製品・企業は、医療機器および体外診断用医療機器に関連する製品を取り扱う企業です。
ここでは、ISO13485の対象となる具体的な製品・企業について解説します。
対象となる医療機器・体外診断用医療機器
適用対象となる製品の定義は以下のとおりです。
- 薬機法で定義される医療機器および体外診断用医療機器(埋め込み型・能動型・診断用機器などを含む)
- 他国の法規制(EU MDR、FDAなど)で規定される医療機器および体外診断用医療機器)
- 上記医療機器に組み込まれる、または機能的に関連する部品、材料、ソフトウェア、装置、試薬、消耗品など
わかりやすいように、医療機器・体外診断用医療機器の具体例をまとめました。自社に関連する製品かどうかを確認する際の参考にしてください。
| 医療機器 | 体外診断用医療機器 |
|---|---|
|
|
対象となる企業
ISO13485を取得できる企業は、医療機器および体外診断用医療機器(IVD)の設計、製造、据付、保守、校正、ならびに使用者への支援を含む関連サービスを提供している組織です。
対象となる企業の具体例をまとめました。
| 医療機器メーカー・製造販売業者 | サプライチェーン関連企業 |
|---|---|
|
|
医療機器は多くの企業によって支えられています。そのため、サプライチェーン関連企業であっても、品質や安全性に影響を与える業務を担っている場合には、ISO認証の取得を求められることがあります。
ISO13485とISO9001・QMS省令の違い

ISO13485と混同しやすい制度に、「ISO9001」と「QMS省令」があります。いずれも品質管理の枠組みという共通点はありますが、対象分野や目的、法的拘束力に大きな違いがあります。
ここでは、ISO13485とISO9001・QMS省令の違いについて解説します。
ISO9001との違い
ISO9001は、業種を問わず導入できる品質マネジメントシステムの国際規格です。目的は、製品・サービスの品質向上と顧客満足の向上にあります。
一方、ISO13485は医療機器分野に特化しており、安全性確保や法規制との整合性を重視している点が大きな違いです。ISO13485は法令遵守やリスク管理の要求が、ISO9001よりも厳しく求められます。
こうした違いから、医療機器メーカーがグローバル展開を目指す場合、ISO9001だけでは不十分となるケースが多いのが実情です。
QMS省令との関係
QMS省令とは、薬機法とISO13485に基づき、日本国内で医療機器を製造・販売するために遵守が義務づけられている法規制です。
ISO13485が国際規格であるのに対し、QMS省令は日本国内向けの法令という位置づけになります。両者は多くの部分で整合しているため、ISO13485の仕組みを構築することが、QMS省令対応にもつながります。
どの規格を取得すべきか?
ISO13485とISO9001のどちらを取得すべきかは、企業の事業内容や将来の展開方針によって異なります。
まずは、ISO13485とISO9001・QMS省令、それぞれの位置づけを整理することが大切です。
| 規格・制度 | 対象 | 目的 | 法的拘束力 |
|---|---|---|---|
| ISO9001 | 業種を問わず適用可能 | 顧客満足・品質の向上 | なし(任意取得) |
| QMS省令 | 日本国内の医療機器・体外診断用医薬品 | 薬機法に基づく安全性確保 | あり(法令) |
| ISO13485 | 医療機器分野(国際的) | 法規制適合と各国の規制との整合性の促進 | なし(任意取得) |
この違いを踏まえると、考え方は次のように整理できます。
- 日本国内で事業を行う場合:QMS省令への対応が前提
- 医療機器分野で国際取引を行う場合:ISO13485の取得が求められることが多い
- 組織全体の品質管理を行いたい場合:ISO9001の取得が有効
つまり、「どれが正解か」ではなく、「自社の事業に何が必要か」という視点で選ぶことが重要です。事業範囲と将来戦略を明確にしたうえで、優先順位を決めて整備していきましょう。
ISO13485の要求事項
ここでは、ISO13485の規格要求事項について、規格の構成とISO13485特有の要求事項について解説します。
規格要求事項の構成
ISO規格は、2012年の改正により「附属書SL(Annex SL)」に基づいた共通構造で要求事項が策定されるようになりました。これにより、ISO9001、ISO14001、ISO/IEC27001などの多くの管理規格が共通の章構成を持つようになっています。
しかし、ISO13485:2016は、医療機器に対する各国規制との整合性を優先する必要があるため、ISO9001:2008をベースにした独自の構成を採用しており、Annex SL形式には準拠していません。そのため、以下のような章構成になっています。
- 0:序文
- 1:適用範囲
- 2:引用規格
- 3:用語及び定義
- 4:品質マネジメントシステム
- 5:経営者の責任
- 6:資源の運用管理
- 7:製品実現
- 8:測定、分析及び改善
- 附属書A
- 附属書B
ISO13485では、自社の製品や業務範囲に直接適用できない要求事項がある場合でも、その適用除外を行うには、明確な根拠と文書化された理由が必要です。単に「関連が薄い」という理由で要求事項への対応を省略することは認められておらず、監査時には適用の妥当性について説明責任が問われることになります。
ISO13485特有の要求事項
本項では、ISO13485:2016の中でも、他のISO規格(例:ISO9001,ISO14001)には見られない、医療機器業界特有の規制対応を反映した要求事項を中心に解説します。
ISO13485は、医療機器の安全性・有効性を担保するため、QMS(品質マネジメントシステム)の観点から、設計、製造、据付、保守、クレーム対応までの全ライフサイクルを対象とした規格であり、以下に示す項目は、各国の医療機器規制とも密接に関係しています。
4.2.3:医療機器ファイルの作成・維持
組織は、各医療機器の型式ごとに、設計・製造・設置・保守・ラベリングなどの情報を体系的にまとめた「医療機器ファイル」を作成・維持する必要があります。このファイルは、製品の要求事項への適合性を文書で示し、各国の規制当局に対する証明資料にもなります。
医療機器ファイルに記載すべき主な内容を以下にまとめました。
- 医療機器の意図する用途や目的、すべての使用説明を含むラベリング・添付文書
- 医療機器の製品仕様・図面
- 製造、保管、取扱い及び配送の仕様または手順
- 測定及び監視手順
- 設置手順
- サービス手順(設置支援、保守など)
6.4:作業環境の衛生管理及び要員の健康や清潔さなどの管理と汚染管理
6.4項は、「6.4.1:作業環境」と「6.4.2:汚染管理」から構成されている項目です。
「6.4.1:作業環境」では、医療機器の品質を確保するために、作業環境を管理することが求められています。そのために、作業環境を監視し、管理するための手順を文書化することが必要です。具体的には、作業員の健康状態や清潔さ、衣類に関する要求事項、医療機器を扱う作業員の力量について規定しています。
「6.4.2:汚染管理」では、医療機器が外部要因(粒子、微生物、化学物質等)によって汚染されることを防ぐため、作業者、作業環境、設備に対して汚染予防計画を策定し、文書化・実施・記録管理が必要です。
※「医療製品の管理」ではなく、「医療機器やそれを扱う環境の汚染制御」のことを指します。
7.5.1:製品及びサービス提供の管理
医療機器および付帯サービス(例:設置・保守)は、意図した使用目的・性能を満たすことが確実になるように、提供前に明確な計画を立て、適切な方法で管理・記録を行うことが求められます。
これには、工程手順の標準化、作業指示書、検査基準、供給者管理なども含まれます。
7.5.2:製品の清浄性及び汚染管理
組織は、製品の使用において清浄性が重要な要因となる場合、医療機器の清浄性および汚染管理に関する手順を確立し、文書化し、実施し、必要に応じて記録する必要があります。特に、以下のいずれかの条件に該当する製品においては、清浄性確保のための制御が求められます。
- 製品を滅菌または使用前に組織によって清浄する場合
- 製品を非滅菌で供給し、滅菌または使用前に清浄する場合
- 製品は滅菌または使用前に清浄できないが、使用時の清浄性が重要である場合
- 製品は滅菌されずに使用されるが、使用時の清浄性が重要である場合
- 製造工程内で製品に残留してはいけない薬剤を使用している場合
また、これらに該当しないと判断する場合でも、その判断の根拠を明確にし、必要に応じてリスク評価等の記録を保持することが求められます。
7.5.5:滅菌医療機器に対するトレーサビリティの確保
滅菌医療機器を製造・供給する場合には、バッチごとの滅菌パラメータ(時間、温度、圧力など)の記録を残し、バッチ単位でのトレースが可能でなければなりません。
製品に問題が発生した場合には、どのバッチに属し、どの条件下で滅菌されたかを迅速に特定することが求められます。このような詳細なロット追跡は、ISO9001では要求されません。
7.5.7:滅菌プロセスなどのバリデーション
組織は、滅菌や無菌バリアシステムなど、完成後に検証が困難または不適切とされる特殊プロセスについて、プロセスバリデーションを実施するための手順を文書化し、実施結果を記録・維持する必要があります。
バリデーションには、以下の内容が含まれなければなりません。
- バリデーションの計画と目的
- 合否判定基準
- 使用機器・装置・環境条件
- 試験方法と結果
- 結果の評価と結論
また、プロセス条件の変更、設備の更新、逸脱の発生時には再バリデーションを実施し、その都度記録を更新する必要があります。
これらの文書は、製品の安全性と有効性を保証する証拠として、規制要求に基づく期間(例:製品寿命+2年など)保管される必要があります。
8.2.3:苦情などの規制当局への報告
医療機器に関する顧客からの苦情や不具合が発生した場合、組織はそれを文書化された手順に従って調査・処置し、必要に応じて各国の規制当局に報告する義務があります。
例えば、日本では以下のような場合に報告義務が生じます。
- 使用者が死亡・重篤な障害を負った場合(医薬品医療機器等法第68条)
- 異物混入・誤使用を引き起こす表示不備があった場合
- リコール等の是正処置を必要とする場合
このような法的報告義務は、ISO9001では存在しないISO13485特有の要求です。
ISO13485要求事項を満たすポイント
ISO13485では品質管理を行うだけでなく、「法令・規制を確実に守れる仕組み」をつくることが求められます。ここでは、特に重要な3つのポイントをわかりやすく整理します。
法令・規制要件の順守を確実にする体制づくり
ISO13485の重要な目的の一つは、「国内外の医療機器関連法規制への適合」です。まず薬機法やEU MDR、FDAなどの規制を洗い出したうえで、自社に関係する法令を整理し、どの製品・業務にどの規制が適用されるのかを明確にします。
そのうえで、以下の取り組みを実施し、法令遵守を確実にする体制づくりを行いましょう。
- 法令管理の責任者を明確にする
- 従業員への教育・訓練を継続的に行う
- 内部監査で運用状況をチェックする
ただし、ISO13485の法令・規制順守は、担当者任せでは達成できません。規制の整理や責任者の設置、教育訓練、内部監査などを通じて、組織全体で仕組みを確実に運用することが重要です。
有効性の高い内部監査の実施
ISO13485の要求事項を満たすためには、内部監査を単なる形式的なチェックにとどめず、規格の要求事項が組織内で確実に運用され、効果的に機能しているかを確認する仕組みとして活用することが重要です。
有効な内部監査を行うには、内部監査員に対する教育訓練で力量を高めることや、必要に応じて外部コンサルタントのサポートを受けることが有効です。
内部監査は、ISO13485の要求事項を満たすだけでなく、PDCAサイクルを回すうえでも欠かせません。内部監査によって潜在的な問題を早期に発見し、改善につなげることで、医療機器の品質と安全性を高めることができます。
苦情対応と是正処置の運用徹底
ISO13485において苦情は、医療機器の品質や安全性に関わる重要な情報源と位置づけられています。単なる顧客の意見や不満ではなく、「潜在的な製品不具合やリスクを把握し、改善につなげるための指標」と考えられます。
そのため、苦情を適切に収集・分析し、迅速かつ効果的に対応することが重要です。具体的には、苦情の内容を記録し、原因を特定したうえで是正処置を実施し、その有効性を確認することが求められます。
ISO13485認証を取得するメリットは?

ISO13485を取得する最大のメリットは、「医療機器に求められる品質と法令遵守を仕組化できる」ことです。
その結果、組織体制の強化はもちろん、顧客や取引先からの信頼向上など、さまざまな面でのメリットへとつながるのです。
ここでは、ISO13485を取得する具体的なメリットをわかりやすく整理します。
製品の品質向上や組織体制の強化につながる
ISO13485を取得するメリットとして、製品のばらつきや人的ミスを低減し、結果として品質の安定や組織的な管理体制の整備につながる点が挙げられます。
ISO13485の「6.資源の運用管理」「7.製品実現」「8.測定、分析及び改善」では、医療機器の安全性および品質の一貫性を確保するために、製造工程の標準化、作業者の力量管理、作業環境の整備、モニタリングなどが求められています。
要求事項を満たす管理体制を構築すれば、作業者による品質の差も最小限に抑えられ、属人化の抑制にも効果的です。
法令遵守を確実にできる
ISO13485は、取得する過程で、設計・製造・販売などの各業務が文書化・記録管理され、法令を守るための体制が整うメリットも見込まれます。同規格では、療機器に関わる法規制への適合を前提とした、品質マネジメントシステムの構築を求めてるためです。
また、ISO13485はISO14971(医療機器リスクマネジメント)との整合性を要求しており、製品のライフサイクル全体におけるリスクの特定・分析・制御が行えるようになります。これにより、重大な不具合や患者リスクの発生を未然に防止する管理体制の構築が可能になります。
調査にかかる手間や費用の低減
ISO13485を取得していると、QMS省令との整合性があると評価され、書類審査がスムーズになるメリットがあります。状況によっては、実地調査の一部が簡略化されることも。日本では、医療機器を扱う企業に対して、薬機法に基づく「QMS適合性調査」が行われます。これは、管理体制がQMS省令に適合しているかを確認する調査です。
ただし、実地調査の最終判断は行政(厚生労働省やPMDA)が個別に判断するため、ISO13485認証があっても免除が確約されるわけではありません。あくまで調査負担が軽減される可能性がある、と考えておきましょう。
顧客からの信頼獲得や新たな顧客獲得につながる
ISO13485認証を取得するということは、「適切に品質を管理できている企業」であることを社外に示せるため、顧客や取引先からの信頼獲得が期待できます。
認証の取得が、自社の品質管理体制が第三者機関(審査機関)により認められた証明になるためです。
欧州のCEマーキングや米国のFDA対応などでは、ISO13485認証が取引条件になる場合もあります。また、国際的な監査制度であるMDSAPもISO13485を基盤としているため、海外展開を目指す際に新たな顧客獲得につながる可能性もあります。
【独自調査】家庭用医療機器に対するニーズ・信頼性に関するアンケート調査結果
実際に、家庭用医療機器を日常的に使用している約1,000人にアンケート調査を行った結果を紹介します。
家庭用医療機器に対するニーズは「安全性・信頼性の高さ」

「安心して使用できる家庭用医療機器にはどのような要素が必要だと思うか」と尋ねたところ、以下のような回答が得られました。
- 厳しい審査基準を通過していて安全性・信頼性が高い(56.5%)
- 長期間、安定して使えるという安心感がある(壊れにくい・性能が安定)(49.4%)
- 使い方が直感的で、誤操作の心配がない(35.0%)
家庭用医療機器の信頼性判断基準には「認証取得」を挙げる声が最多
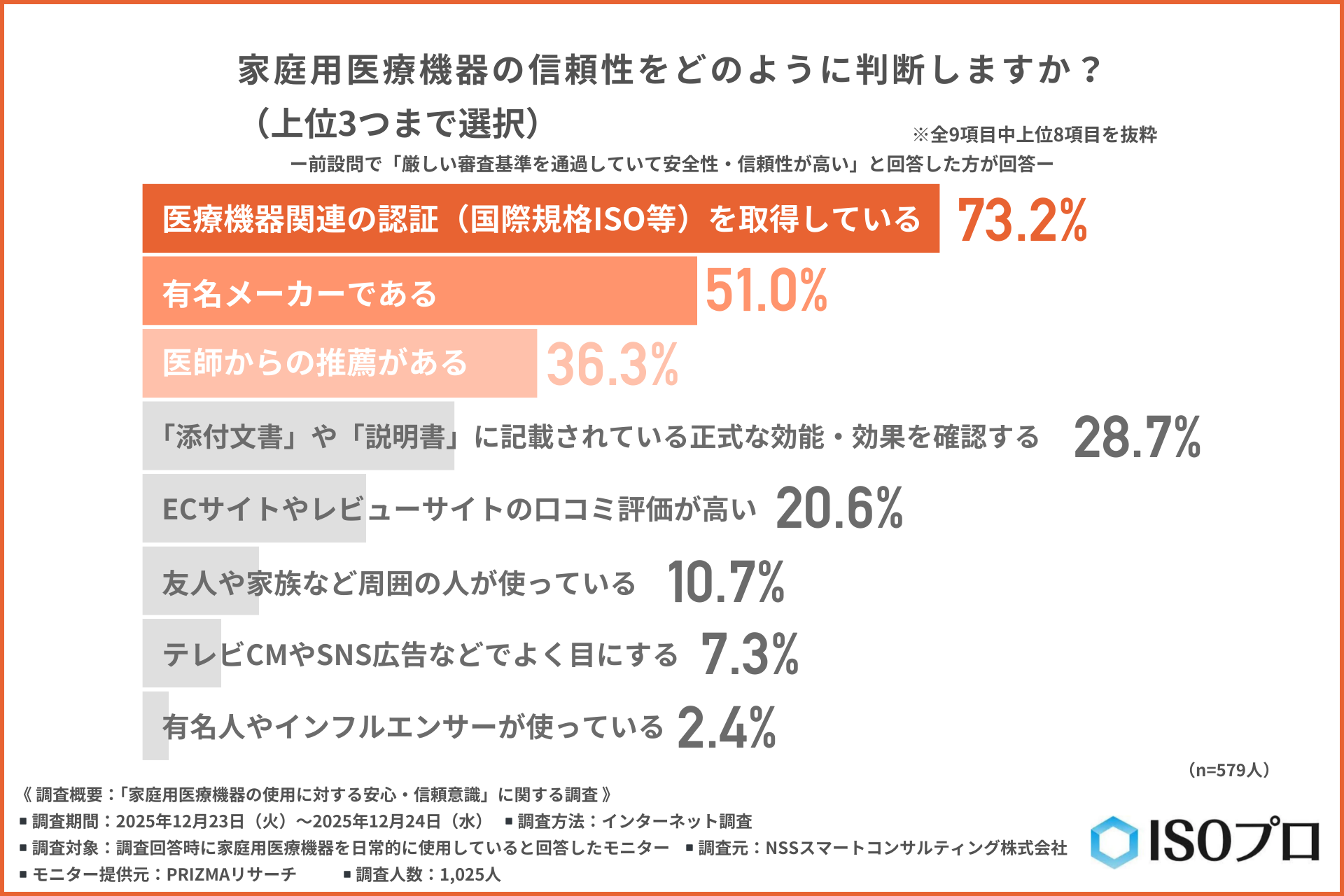
『厳しい審査基準を通過していて安全性・信頼性が高い』と回答した方に、さらに以下の質問を行いました。
「家庭用医療機器の信頼性をどのように判断するか」について尋ねたところ、以下のような回答結果となりました。
- 医療機器関連の認証(ISOなど)を取得している(73.2%)
- 有名メーカーである(51.0%)
- 医師からの推薦がある(36.3%)
この結果から、顧客からの信頼を得るためには「医療機器関連からの第三者認証」が最も強力な根拠となっていることがわかりました。
ISO13485の取得プロセスと所要期間
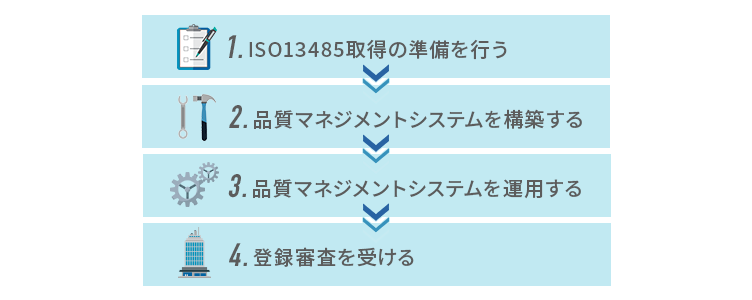
ISO13485を取得するには、準備から運用、認証審査、取得までを計画的に進める必要があります。取得にかかる期間は組織の規模や体制によりますが、一般的には約6か月~12か月の時間がかかります。
ここでは、ISO13485を取得する流れを解説します。
1.ISO13485取得の準備を行う
まずはISO13485取得の準備を行いましょう。
取得準備に際しては、まず自社が扱う医療機器の分類(クラスI~III/IV)、事業スコープ(製造/販売/設計等)を明確にし、ISO13485が要求する品質マネジメントの適用範囲を決定することが重要です。
取得に向けたプロジェクトチームを発足するとともに、自社だけで取得を目指すのか、コンサルに取得サポートを依頼するのかどうかを決定します。
ISO規格の取得に関するノウハウがない場合には、プロのコンサルに依頼することで取得にかかる自社の工数や期間、コストの削減につながります。
2.品質マネジメントシステムを構築する
要求事項を満たす品質マネジメントシステムを構築します。
その際には自社の現在の取り組みや品質管理体制を、ISO13485の規格要求事項と照らし合わせて、不足している部分を明確にすることが大切です。
自社の業務プロセスとISO13485の要求事項との間にある差異を文書・運用・記録の観点から評価し、リスクマネジメント、バリデーション、文書管理など医療機器特有の要件が確実に盛り込まれるよう整備する必要があります。
3.品質マネジメントシステムを運用する
品質マネジメントシステムを構築したら、実際に運用します。運用開始後は、内部監査とマネジメントレビューを少なくとも1回以上実施し、システムの有効性をトップマネジメントが評価した記録を残す必要があります。
4.登録審査を受ける
登録審査は、通常2段階で構成されます。
- 第一段階審査(Stage1)では、品質マニュアルや主要文書の整合性、申請範囲、運用記録の有無などを確認します。
- 第二段階審査(Stage2)では、実際の運用現場において、文書と実務が整合しているか、トレーサビリティ・記録・力量管理などがISO13485に従って実施されているかを審査員が現場確認とインタビューを通じて評価します。
この2段階審査を通過し、不適合が是正されたことが確認されると、ISO13485認証が付与されます。
5.継続的な運用と認証維持
ISO13485取得後も、管理体制を維持するには継続的な運用が欠かせません。定期的な内部監査やマネジメントレビューを行い、改善策の実施状況を確認することで、マネジメントシステムを常に有効に機能させる必要があります。
また、外部審査機関による定期的な維持審査・更新審査を受けることが求められます。規格に適合しているかどうかが確認されます。
ISO13485に関するよくある質問
ISO13485に関するよくある質問をQ&A形式でまとめました。
Q:ISO13485はISO9001と何が違う?
ISO13485は「医療機器業界に特化した品質マネジメント規格」、ISO9001は「あらゆる業種に使える一般的な品質マネジメント規格」です。
ISO13485は、医療機器の安全性確保や法規制対応を重視しており、要求がより厳格に定められています。医療機器分野では、ISO9001よりもISO13485の方が実務に直結します。
Q:ISO13485取得は必須?
いいえ、日本国内で医療機器の製造販売を行う場合、ISO13485取得は法律上の義務ではありません。
ただし、海外取引や大手企業との取引では、取得が事実上の条件になるケースもあります。特に欧州や北米市場を目指す場合は、取得しておくことが有利です。
Q:ISO13485取得にはどれくらいの期間と費用がかかる?
一般的にかかる期間は、準備から認証取得まで6か月〜1年程度が目安です。
費用相場は、コンサル費用・審査費用などを含めて100万円~300万円程度が一つの目安です。
ただし、体制の整備状況や対象製品の範囲によって大きく変わるため、事前の見積もり確認が重要です。
まとめ
ISO13485は、医療機器に関する国際的な品質マネジメントシステムの規格であり、ISO9001をベースとしつつも、医療機器の安全性・有効性を担保するために必要なプロセスの厳格な管理を求める内容になっています。
この規格に基づいたマネジメントシステムを構築・運用することで、自社の法令遵守体制の強化、製品安全リスクの軽減、取引先・審査機関への信頼性向上といった多面的な効果が期待されます。
ISOプロでは月額4万円から御社に合わせたISO運用を実施中
ISOプロではISO各種の認証取得から運用まで幅広くサポートしております。
また、マニュアル作成など御社に合わせたムダのない運用を心がけており、既に認証を取得しているお客様においてもご提案しております。
サポート料金においても新プランを用意し、業界最安級の月額4万円からご利用いただけます。


































こんな方に読んでほしい